
4 Marzo 2018. Il capitano della Fiorentina Davide Astori viene trovato privo di vita nella sua stanza d’albergo a Udine, dove si era recato con la sua squadra per giocare la partita di campionato Udinese – Fiorentina. Nelle settimane seguenti, in molti hanno provato a dare una spiegazione alla morte improvvisa del giocatore suggerendo la Sindrome di Brugada come possibile causa. La realtà è che ancora troppo presto per capire cosa abbia causato l’arresto cardiaco di Astori e bisognerà aspettare l’esito di esami più approfonditi. Considerato il clamore mediatico dato alla Sindrome di Brugada, vediamo di capire un po meglio di cosa si tratta.
La Sindrome di Brugada venne descritta per la prima volta dai fratelli Pedro e Josep Brugada che mostrarono l’associazione tra un pattern elettrocardiografico tipico e morte improvvisa o arresto cardiaco per fibrillazione ventricolare. Questo aspetto all’ECG a riposo (definito Pattern di tipo I) consiste nell’ innalzamento del punto J (ovvero il punto che corrisponde alla fine del complesso QRS, che rappresenta l’attivazione elettrica dei ventricoli cardiaci) > o = a 2mm, nel sopraslivellamento discendente del tratto ST e in una successiva onda T negativa (entrambi aspetti dell’ECG a riposo correlati con i fenomeni di ripolarizzazione dei ventricoli, ovvero di ritorno al loro riposo elettrico). Successivamente vennero riscontrati altri due aspetti tipici della sindrome: il Pattern tipo 2 (che si differenzia dal Tipo 1 per il sopraslivellamento del tratto ST a plateau a cui segue un onda T positiva) e il Tipo 3 (che racchiude gli aspetti ECG simili al Tipo 1 e 2 ma che non soddisfano in pieno i criteri diagnostici). (Vedi figura).
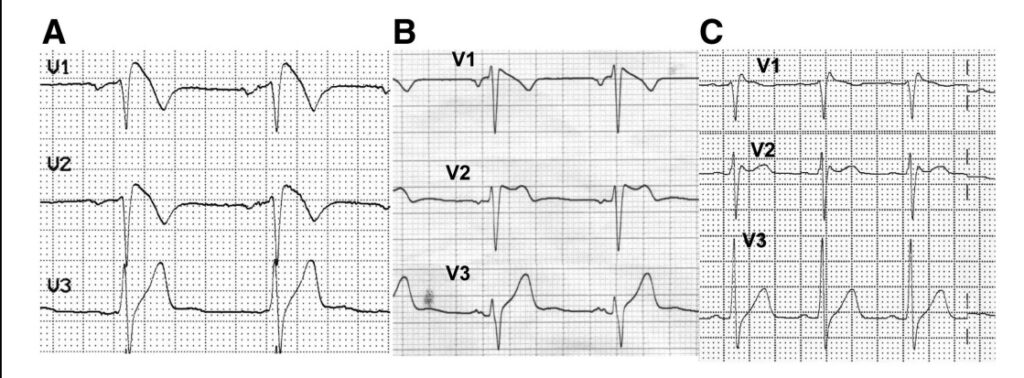
Figura 1. A – Pattern Brugada tipo 1. B – Pattern Brugada tipo 2. C – Pattern Brugada tipo 3. Da Berne P e Brugada J. Brugada Syndrome 2012. Circulation Journal Vol 26 July 2012.
Dal momento della sua prima descrizione, la Sindrome di Brugada è stata ampiamente studiata ed è ancora in fase di studio non essendo stati elucidati del tutto i meccanismi ad essa legati. Al momento si sa che è una patologia legata alla mutazione di geni coinvolti nella produzione di proteine che fungono da canali ionici nelle cellule cardiache, che quindi hanno un ruolo importante nella genesi e conduzione elettrica nel cuore. La mutazione più frequentemente associata alla Sindrome è a carico del gene SCNA5 che contiene le informazioni per la produzione di un canale ionico del Sodio, ma sono associate alla patologia anche mutazioni a carico di geni coinvolti nella produzione di canali ionici per il Potassio, il Calcio nonché di geni coinvolti nella sintesi di proteine del desmosoma, ovvero la struttura proteica che tiene unite le cellule cardiache e ne permette l’attività meccanica ed elettrica coordinata.
La malattia esordisce maggiormente in età comprese tra i 40 e i 45 anni, ma può mostrare i propri segni ECG o i primi sintomi anche in altre fasce di età. Il sesso maschile è maggiormente coinvolto nella patologia, verosimilmente per l’effetto che hanno gli estrogeni e il testosterone sulla sintesi proteica dei canali ionici cardiaci.
Spesso la Sindrome di Brugada è asintomatica e il percorso diagnostico parte dal riscontro accidentale dell’aspetto ECG tipico durante valutazioni di screening come la visita sportiva. Talvolta la malattia viene scoperta in seguito ad accertamenti per svenimenti immotivati (sincope) e palpitazioni; purtroppo in casi più sfortunati, la malattia esordisce con l’arresto cardiaco e la morte improvvisa associato alla Fibrillazione Ventricolare, spesso durante il sonno o situazioni di riposo a differenza di altre patologie che manifestano questi gravi sintomi durante esercizio. La diagnosi si basa sul riscontro del Pattern Tipo 1 all’ECG a riposo o in seguito alla somministrazione di alcuni farmaci antiaritmici (come l’ajmailina o la flecainide) durante test farmacologico in pazienti con Pattern di tipo 2 o 3 e sintomi quali sincope, palpitazioni o arresto cardiaco resuscitato. In alcuni casi può essere utile eseguire uno studio elettrofisiologico con l’obiettivo di indurre aritmie maligne nel paziente per verificare la vulnerabilità del tessuto cardiaco; uno studio elettrofisiologico negativo è indice di basso rischio, ma la sua positività non conferma la presenza della vulnerabilità e per questo motivo non sempre viene praticato.
Il trattamento consiste nell’impianto del Defibrillatore Impiantabile nei soggetti più a rischio (ovvero chi ha già avuto un arresto cardiaco o episodi di Tachicardia Ventricolare, in soggetti con storia di sincope) o nell’uso di farmaci antiaritmici come la chinidina. Al momento i soggetti asintomatici considerati a basso rischio vengono seguiti nel tempo consigliando loro di evitare le grandi abbuffate, l’alcool, l’uso di certi farmaci (per una lista aggiornata visitare il sito www.brugadadrugs.org) e di controllare prontamente con antipiretici la febbre, che può scatenare aritmie maligne nei soggetti affetti.
Per quanto riguarda l’idoneità sportiva agonistica, purtroppo la sindrome di Brugada al momento è da considerarsi motivo di non idoneità allo sport con l’eccezione di atleti portatori di Pattern tipo 2 o 3 asintomatici e privi di familiarità per morte improvvisa. L’idonietà può essere presa in considerazione anche in portatori di Pattern di tipo 1 ma asintomatici, con studio elettrofisiologico negativo, in assenza di storia familiare per morte improvvisa e di fattori di rischio minori.
Bibliografia:
- Berne P e Brugada J. Brugada Syndrome 2012. Circulation Journal Vol 26 July 2012;
- Siera J and Brugada P. The definition of Brugada Syndrome. European Heart Journal (2017) 38, 3029–3034;
- Priori SG et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC) Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). European- Heart Journal, Volume 36, Issue 41, 1 November 2015, Pages 2793–2867.
- Protocolli cardiologici per il giudizio di idoneità agonistico 2017 (COCIS).